|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Anesthetics cause dose-related and reversible alterations in many aspects of cerebral physiology, including CBF, cerebral metabolic rate (CMR), and electrophysiologic function (EEG, evoked responses). The effects of anesthetic drugs and techniques have the potential to adversely affect the diseased brain and performance of the neurosurgical procedure and are therefore of clinical importance in patients with neurologic disease. However, in certain instances, the effects of general anesthesia on CBF and CMR can be manipulated to improve both the operative course and the clinical outcome of patients with neurologic disorders.
The adult human brain weighs approximately 1350 g and therefore represents about 2% of total-body weight. However, it receives 12% to 15% of cardiac output. This high flow rate is a reflection of the brain's high metabolic rate. At rest, the brain consumes oxygen at an average rate of approximately 3.5 mL of oxygen per 100 g of brain tissue per minute. Whole-brain O2 consumption (13.5 × 3.5 = 47 mL/min) represents about 20% of total-body oxygen utilization. Normal values for CBF,
| CBF |
|
| Global | 45–55 mL/100 g/min |
| Cortical (mostly gray matter) | 75–80 mL/100 g/min |
| Subcortical (mostly white matter) | ∼20 mL/100 g/min |
| CMRO2 | 3–3.5 mL/100 g/min |
| CVR | 1.5–2.1 mm Hg/100 g/min/mL |
| Cerebral venous PO2 | 32–44 mm Hg |
| Cerebral venous SO2 | 55%–70% |
| ICP (supine) | 8–12 mm Hg |
| CBF, cerebral blood flow; CMRO2 , cerebral metabolic rate of oxygen; CVR, cerebral vascular resistance; ICP, intracranial pressure. | |
Approximately 60% of the brain's energy consumption[1] is used to support electrophysiologic function. The depolarization-repolarization activity that occurs and is reflected in the EEG requires energy expenditure for the maintenance and restoration of ionic gradients and for the synthesis, transport, and reuptake of neurotransmitters. The remainder of the energy consumed by the brain is involved in cellular homeostatic activities. Local CBF and local CMR within the brain are very heterogeneous, and both are approximately four times greater in gray matter than white matter. The cell population of the brain is also heterogeneous in its oxygen requirements. Glial cells make up about half the brain's volume and require less energy than neurons. Besides providing a physically supportive latticework for the brain, glial cells are important in the reuptake of neurotransmitters and the delivery and removal of metabolic substrates and wastes.
The brain's substantial demand for substrate must be met by adequate delivery of oxygen and glucose. However, the space constraints imposed by the noncompliant cranium and meninges require that blood flow not be excessive. Not surprisingly, there are elaborate mechanisms for the regulation of CBF. These mechanisms, which include chemical, myogenic, and neurogenic factors, are listed in Table 21-2 . The precise mechanisms of these effects are for the most part not well understood. However, a substantial volume of largely recent research indicates that modulation of the arginine-nitric oxide (NO)-cyclic guanosine monophosphate system[2] [3] is central to the changes in cerebral vascular tone caused by several processes, including hypercapnia,[4] [5] increased CMR,[6] [7] volatile anesthetics,[8] [9] [10] and neurogenic mechanisms.[11] [12] [13]
Several factors cause changes in the cerebral biochemical environment that result in adjustments in CBF, including changes in CMR, PaCO2 , and PaO2 .
Increased neuronal activity results in increased local brain metabolism,
and this increase in CMR is associated
| Factor | Comment |
|---|---|
| Chemical/Metabolic/Humoral | |
| Cerebral metabolic rate (CMR) | CMR influence assumes intact flow-metabolism coupling, the mechanism of which is not fully understood |
| Anesthetics |
|
| Temperature |
|
| Arousal/seizures |
|
| PaCO2 |
|
| PaO2 |
|
| Vasoactive drugs |
|
| Anesthetics |
|
| Vasodilators |
|
| Vasopressors |
|
| Myogenic | |
| Autoregulation/mean arterial pressure | The autoregulation mechanism is fragile, and in many pathologic states cerebral blood flow is regionally pressure passive |
| Rheologic | |
| Blood viscosity |
|
| Neurogenic | |
| Extracranial sympathetic and parasympathetic pathways | Contribution and clinical significance poorly defined |
| Intra-axial pathways |
|
CMR is influenced by several phenomena in the neurosurgical environment, including the functional state of the nervous system, anesthetics, and temperature.
CMR decreases during sleep and increases during sensory stimulation, mental tasks, or arousal of any cause. During epileptic activity, CMR increases may be extreme, whereas in coma, CMR may be substantially reduced.
The effect of individual anesthetics on CMR is presented in greater detail later in this chapter in the section "Effects of Anesthetics on Cerebral Blood Flow and Cerebral Metabolic Rate." In general, anesthetics suppress CMR, with ketamine and nitrous oxide (N2 O) being notable exceptions. It appears that the component of CMR on which they act is that associated with electrophysiologic function. [21] With several anesthetics, including barbiturates, [21] isoflurane,[22] sevoflurane,[23] desflurane,[24] propofol,[25] and etomidate,[26] increasing plasma concentrations cause progressive suppression of EEG activity and a concomitant reduction in CMR. However, increasing the plasma level beyond what is required to first achieve suppression of the EEG results in no further depression of CMR. The component of CMR required for maintenance of cellular integrity, the "housekeeping" component, is apparently unaltered by intravenous anesthetics ( Fig. 21-1 ).
Values of the cerebral metabolic rate of oxygen (CMRO2 ) observed when complete suppression of the EEG is achieved with different anesthetics are very similar. The inference that anesthetic-induced EEG suppression
 Figure 21-1
The interdependency[21]
of cerebral electrophysiologic function and cerebral metabolic rate (CMR). Administration
of various anesthetics,[21]
[22]
[26]
including barbiturates, results in a dose-related
reduction in the cerebral metabolic rate of oxygen (CMRO2
) and cerebral
blood flow (CBF). The maximum reduction occurs with the dose that results in electrophysiologic
silence. At this point, energy utilization associated with electrophysiologic activity
has been reduced to zero, but energy utilization for cellular homeostasis persists
unchanged. Additional barbiturate causes no further decrease in CBF or CMRO2
.
EEG, electroencephalogram.
Figure 21-1
The interdependency[21]
of cerebral electrophysiologic function and cerebral metabolic rate (CMR). Administration
of various anesthetics,[21]
[22]
[26]
including barbiturates, results in a dose-related
reduction in the cerebral metabolic rate of oxygen (CMRO2
) and cerebral
blood flow (CBF). The maximum reduction occurs with the dose that results in electrophysiologic
silence. At this point, energy utilization associated with electrophysiologic activity
has been reduced to zero, but energy utilization for cellular homeostasis persists
unchanged. Additional barbiturate causes no further decrease in CBF or CMRO2
.
EEG, electroencephalogram.
 Figure 21-2
Cortical somatosensory evoked responses to median nerve
stimulation in humans before induction and during anesthesia with thiopental and
isoflurane/N2
O. In spite of an equivalent or greater degree of reduction
in cerebral metabolic rate with thiopental, cortical evoked responses are better
preserved[30]
than during anesthesia with isoflurane,
[31]
which suggests that the electroencephalographic
suppression achieved with different anesthetic agents should not be assumed to be
equivalent electrophysiologic states. The cumulative thiopental doses and expired
concentrations of isoflurane and N2
O are indicated.
Figure 21-2
Cortical somatosensory evoked responses to median nerve
stimulation in humans before induction and during anesthesia with thiopental and
isoflurane/N2
O. In spite of an equivalent or greater degree of reduction
in cerebral metabolic rate with thiopental, cortical evoked responses are better
preserved[30]
than during anesthesia with isoflurane,
[31]
which suggests that the electroencephalographic
suppression achieved with different anesthetic agents should not be assumed to be
equivalent electrophysiologic states. The cumulative thiopental doses and expired
concentrations of isoflurane and N2
O are indicated.
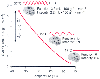 Figure 21-3
The effect of temperature reduction on the cerebral metabolic
rate of oxygen (CMRO2
). Hypothermia reduces both of the components of
cerebral metabolic activity identified in Figure
21-1
: that associated with neuronal electrophysiologic activity ("Function")
and that associated with maintenance of cellular homeostasis ("Integrity"). This
effect is in contrast to anesthetics that alter only the functional component. The
ratio of CMR at 37°C to that at 27°C, the Q10
ratio, is shown
in the graph. EEG, electroencephalogram. (Adapted from Michenfelder JD:
Anesthesia and the Brain: Clinical, Functional, Metabolic, and Vascular Correlates.
New York, Churchill Livingstone, 1988.)
Figure 21-3
The effect of temperature reduction on the cerebral metabolic
rate of oxygen (CMRO2
). Hypothermia reduces both of the components of
cerebral metabolic activity identified in Figure
21-1
: that associated with neuronal electrophysiologic activity ("Function")
and that associated with maintenance of cellular homeostasis ("Integrity"). This
effect is in contrast to anesthetics that alter only the functional component. The
ratio of CMR at 37°C to that at 27°C, the Q10
ratio, is shown
in the graph. EEG, electroencephalogram. (Adapted from Michenfelder JD:
Anesthesia and the Brain: Clinical, Functional, Metabolic, and Vascular Correlates.
New York, Churchill Livingstone, 1988.)
The effects of hypothermia on both normal and ischemic brain have been reviewed in detail.[1] [33] [34] CMR decreases by 6% to 7%/°C of temperature reduction.[1] As is the case with some anesthetics, hypothermia can also cause complete suppression of the EEG (at about 18°C to 20°C). However, in contrast to anesthetics, temperature reduction beyond that at which EEG suppression first occurs does produce a further decrease in CMR ( Fig. 21-3 ). This decrease occurs because although anesthetics reduce only the component of CMR associated with neuronal function, hypothermia causes decreases in the rate of energy utilization associated with both electrophysiologic function and the basal component associated with maintenance of cellular integrity. These decreases were once assumed to be proportional. However, mild hypothermia preferentially suppresses the basal component of CMR.[35] [36] CMRO2 at 18°C is less than 10% of normothermic control values, which probably accounts for the brain's tolerance of moderate periods of circulatory arrest at these and lower temperatures.
Hyperthermia has an opposite influence on cerebral physiology. Between 37°C and 42°C, CBF and CMR increase.[37] However, above 42°C a dramatic reduction in cerebral oxygen consumption occurs, an indication of a threshold for a toxic effect of hyperthermia that may occur as a result of protein (enzyme) degradation.
CBF varies directly with PaCO2 ( Fig. 21-4 ). The effect is greatest within the range of physiologic PaCO2 variation. CBF changes 1 to 2 mL/100 g/min for each 1-mm Hg change in PaCO2 around normal PaCO2 values.[38] This
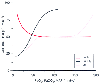 Figure 21-4
Changes in cerebral blood flow (CBF) caused by independent
alterations in PaCO2
, PaO2
,
and mean arterial pressure (MAP).
Figure 21-4
Changes in cerebral blood flow (CBF) caused by independent
alterations in PaCO2
, PaO2
,
and mean arterial pressure (MAP).
The changes in CBF caused by PaCO2 are apparently dependent on pH alterations in the extracellular fluid of the brain. [40] NO, especially NO of neuronal origin, is an important, though not exclusive mediator of CO2 -induced vasodilatation. [4] [5] [41] [42] [43] In particular, in primates, NO's relevance may be limited to the cerebral cortex.[5] In humans, NO inhibition significantly attenuates the hyperemic response to hypercapnia. [44] The vasodilator response to hypercapnia is also mediated in part by prostaglandins; administration of indomethacin reduces the vasodilator response by about 60%.[45] The changes in extracellular pH and CBF occur rapidly after PaCO2 adjustments because CO2 diffuses freely across the cerebrovascular endothelium. Note that in contrast to respiratory acidosis, acute systemic metabolic acidosis has little immediate effect on CBF because the blood-brain barrier (BBB) excludes the hydrogen ion (H+ ) from the perivascular space. Although the CBF changes in response to alterations in PaCO2 occur rapidly, they are not sustained. In spite of the maintenance of increased arterial pH, CBF returns to normal over a period of 6 to 8 hours[46] [47] [48] because cerebrospinal fluid (CSF) pH gradually returns to normal as a result of the extrusion of bicarbonate. Consequently, a patient who has had a sustained period of hyperventilation or hypoventilation
Changes in PaO2 from 60 to over 300 mm Hg have little influence on CBF. Below a PaO2 of 60 mm Hg, however, CBF increases rapidly (see Fig. 21-4 ). The mechanisms mediating the cerebral vasodilation during hypoxia are not fully understood but may include neurogenic effects initiated by peripheral or neuraxial chemoreceptors, as well as local humoral influences. At least part of the hyperemic response to hypoxia is mediated by NO of neuronal origin.[49] Hypoxia-induced hyperpolarization of vascular smooth muscle by the opening of adenosine triphosphate (ATP)-dependent K+ channels also leads to vasodilation. Recent studies have indicated that the rostral ventrolateral medulla (RVM) serves as an oxygen sensor within the brain.[50] Stimulation of the RVM by hypoxia results in an increase in CBF (but not CMR), and lesions of the RVM suppress the magnitude of the CBF response to hypoxia. The response to hypoxia is synergistic with the hyperemia produced by hypercapnia and acidosis. At high PaO2 values, CBF decreases modestly.[51] At 1 atm of oxygen, CBF is reduced by 12%.[52]
|
|
|
|
|
|
|
|
|
|
|
|
|