|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
The pulmonary vascular bed is a high-flow, low-pressure system
in health. As ![]() T increases, pulmonary vascular
pressures increase minimally.[18]
However, increases
in
T increases, pulmonary vascular
pressures increase minimally.[18]
However, increases
in ![]() T distend open vessels and recruit previously
closed vessels.
T distend open vessels and recruit previously
closed vessels.
 Figure 17-7
Passive changes in pulmonary vascular resistance (PVR)
as a function of pulmonary artery pressure (Ppa) and pulmonary blood flow (
Figure 17-7
Passive changes in pulmonary vascular resistance (PVR)
as a function of pulmonary artery pressure (Ppa) and pulmonary blood flow (![]() T)
(PVR = Ppa/QT). As
T)
(PVR = Ppa/QT). As ![]() T
increases, Ppa also increases, but to a lesser extent, and PVR decreases. As QT
decreases, Ppa also decreases, but to a lesser extent, and PVR increases. (Redrawn
with modification from Fishman AP: Dynamics of the pulmonary circulation. In
Hamilton WF [ed]: Handbook of Physiology, section 2. Circulation, vol 2. Baltimore,
Williams & Wilkins, 1963, p 1667.)
T
increases, Ppa also increases, but to a lesser extent, and PVR decreases. As QT
decreases, Ppa also decreases, but to a lesser extent, and PVR increases. (Redrawn
with modification from Fishman AP: Dynamics of the pulmonary circulation. In
Hamilton WF [ed]: Handbook of Physiology, section 2. Circulation, vol 2. Baltimore,
Williams & Wilkins, 1963, p 1667.)
Understanding the relationship among Ppa, PVR, and ![]() T
during passive events is a prerequisite to recognition of active vasomotion in the
pulmonary circulation (see the next section). Active vasoconstriction occurs whenever
T
during passive events is a prerequisite to recognition of active vasomotion in the
pulmonary circulation (see the next section). Active vasoconstriction occurs whenever
![]() T decreases and Ppa remains either constant
or increases. Increased Ppa and PVR have been found to be "a universal feature of
acute respiratory failure."[19]
Active pulmonary
vasoconstriction can increase Ppa and Ppv, thereby contributing to the formation
of pulmonary edema, and in that way has a role in the genesis of adult respiratory
distress syndrome (ARDS). Active vasodilation occurs whenever
T decreases and Ppa remains either constant
or increases. Increased Ppa and PVR have been found to be "a universal feature of
acute respiratory failure."[19]
Active pulmonary
vasoconstriction can increase Ppa and Ppv, thereby contributing to the formation
of pulmonary edema, and in that way has a role in the genesis of adult respiratory
distress syndrome (ARDS). Active vasodilation occurs whenever ![]() T
increases and Ppa either remains constant or decreases. When deliberate hypotension
is achieved with sodium nitroprusside,
T
increases and Ppa either remains constant or decreases. When deliberate hypotension
is achieved with sodium nitroprusside, ![]() T often
remains constant or increases, but Ppa decreases, and therefore so does PVR.
T often
remains constant or increases, but Ppa decreases, and therefore so does PVR.
Lung volume and PVR have an asymmetric U-shaped relationship because of the varying effect of lung volume on intra- and extra-alveolar vessels, which in both cases is minimal at functional residual capacity (FRC). FRC is defined as the amount of volume (gas) in the lungs at end-exhalation during normal tidal breathing. Ideally, this means that the patient is inspiring a normal VT, with minimal or no muscle activity or pressure difference between the alveoli and atmosphere at end-exhalation. Total PVR is increased when lung volume is either increased or decreased from FRC[20] [21] [22] ( Fig. 17-8 ). The increase in total PVR above FRC is due to alveolar compression of small intra-alveolar vessels, which results in an increase in small-vessel PVR (i.e., creation of zone 1 or zone 2).[24] As a relatively small mitigating or counterbalancing effect to the compression of small vessels, the large extra-alveolar vessels may be expanded by the increased tethering of interstitial connective tissue at high lung volumes (and with spontaneous ventilation only—the negativity of perivascular pressure at high lung volumes). The increase in total PVR below FRC is due to an increase in the PVR of large extra-alveolar vessels (passive effect). The increase in large-vessel PVR is partly due to mechanical tortuosity or kinking of these vessels (passive effect). However, small or grossly atelectatic lungs become hypoxic, and it
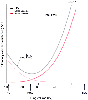 Figure 17-8
Total pulmonary vascular resistance relates to lung volume
as an asymmetric U-shaped curve. The trough of the curve occurs when lung volume
equals functional residual capacity (FRC). Total pulmonary resistance is the sum
of the resistance in small vessels (increased by increasing lung volume) and the
resistance in large vessels (increased by decreasing lung volume). The end point
for increasing lung volume (toward total lung capacity [TLC]) is the creation of
zone 1 conditions, and the end point for decreasing lung volume (toward residual
volume [RV]) is the creation of low ventilation-perfusion (V̇A/
Figure 17-8
Total pulmonary vascular resistance relates to lung volume
as an asymmetric U-shaped curve. The trough of the curve occurs when lung volume
equals functional residual capacity (FRC). Total pulmonary resistance is the sum
of the resistance in small vessels (increased by increasing lung volume) and the
resistance in large vessels (increased by decreasing lung volume). The end point
for increasing lung volume (toward total lung capacity [TLC]) is the creation of
zone 1 conditions, and the end point for decreasing lung volume (toward residual
volume [RV]) is the creation of low ventilation-perfusion (V̇A/![]() )
and atelectatic (atel) areas that demonstrate hypoxic pulmonary vasoconstriction
(HPV). The curve represents a composite of data from references [20]
[21]
and [23]
.
)
and atelectatic (atel) areas that demonstrate hypoxic pulmonary vasoconstriction
(HPV). The curve represents a composite of data from references [20]
[21]
and [23]
.
Four major categories of active processes affect the pulmonary vascular tone of normal patients: (1) local tissue (endothelial and smooth muscle)-derived autocrine/paracrine products, which act on smooth muscle ( Table 17-1 ); (2) alveolar gas concentrations (chiefly hypoxia), which also act on smooth muscle; (3) neural influences; and (4) humoral (or hormonal) effects of circulating products within the pulmonary capillary bed. The neural and humoral effects work by means of either receptor-mediated mechanisms involving the autocrine/paracrine molecules listed in Table 17-1 or related mechanisms ultimately affecting the smooth muscle cell.[26] These four interrelated systems, each affecting pulmonary vascular tone, will be briefly reviewed in sequence.
The pulmonary vascular endothelium synthesizes, metabolizes, and converts a multitude of vasoactive mediators and plays a central role in the regulation of PVR. However, the main effecter site of pulmonary vascular tone is the pulmonary vascular smooth muscle cell (which both senses and produces multiple pulmonary vasoactive compounds).[27] The autocrine/paracrine molecules listed in Table 17-1 are all actively involved in the regulation of pulmonary vascular tone during various conditions. Numerous additional compounds bind to receptors on the endothelial or smooth muscle cell membranes and modulate the levels (and effects) of these vasoactive molecules.
Nitric oxide (NO) is the predominant (but not the only) endogenous
vasodilatory compound. It was discovered to be the long-sought-after endothelial-derived
relaxant factor (EDRF) over a decade ago by Moncada and Palmer.[28]
Since then, a massive amount of laboratory and clinical research has demonstrated
the ubiquitous nature of NO and its predominant role in vasodilation of both pulmonary
and systemic blood vessels.[29]
In the pulmonary
endothelial cell, L-arginine is converted to L-citrulline
| Molecule | Subtype (Abbreviation) | Site of Origin | Site of Action | Response |
|---|---|---|---|---|
| Nitric oxide | NO | Endothelium | Sm. muscle | Vasodilation |
| Endothelin | ET-1 | Endothelium | Sm. muscle (ETA receptor) | Vasoconstriction |
|
|
ET-1 | Endothelium | Endothelium (ETB receptor) | Vasodilation |
| Prostaglandin | PGI2 | Endothelium | Endothelium | Vasodilation |
| Prostaglandin | PGF2 α | Endothelium | Sm. muscle | Vasoconstriction |
| Thromboxane | TXA2 | Endothelium | Sm. muscle | Vasoconstriction |
| Leukotriene | LTB4 -LTE4 | Endothelium | Sm. muscle | Vasoconstriction |
| ETA receptor, endothelin-1 receptor located on the smooth muscle cell membrane; ETB receptor, endothelin-1 receptor located on the endothelial cell membrane; Sm. muscle, pulmonary arteriole smooth muscle cell. | ||||
Endothelin-1 (ET-1) is a recently discovered pulmonary vasoconstrictor. [33] The endothelins are 21-amino acid peptides produced by a variety of cells. ET-1 is the only family member produced in pulmonary endothelial cells, and it is also produced in vascular smooth muscle cells.[33] ET-1 exerts its major vascular effects through activation of two distinct G protein-coupled receptors (ETA and ETB). ETA receptors are found in the medial smooth muscle layers of the pulmonary (and systemic) blood vessels and in atrial and ventricular myocardium. [33] When stimulated, ETA receptors induce vasoconstriction and cellular proliferation by increasing intracellular calcium.[34] ETB receptors are localized on endothelial cells and some smooth muscle cells.[35] Activation of ETB receptors stimulates the release of NO and prostacyclin, thereby promoting pulmonary vasodilation and inhibiting apoptosis.[36] Additionally, the ET-1 receptor antagonist bosentan showed modest improvement in the treatment of pulmonary hypertension.[37] Recently, the more selective ETA receptor antagonist sitaxsentan showed additional benefit in improving pulmonary hypertension.[38] However, both ET-1 receptor antagonists (bosentan and sitaxsentan) are associated with an increased risk of liver toxicity.[39] In summary, it appears that there is a normal
Similarly, various eicosanoids are elaborated by the pulmonary vascular endothelium, with a balance toward the vasodilatory compounds in health. Prostaglandin I2 (PGI2 ), now known as epoprostenol (previously known as prostacyclin), causes vasodilation and is continuously elaborated in small amounts in healthy endothelium. In contrast, thromboxane A2 and leukotriene B4 are elaborated under pathologic conditions and are thought to be involved in the pathophysiology of pulmonary artery hypertension (PAH) associated with sepsis and reperfusion injury.[40]
Therapeutically, epoprostenol has been successfully used to decrease PVR in patients with chronic PAH when infused[41] or inhaled.[42] Currently, the synthetic PGI2 iloprost is now the most commonly used inhaled eicosanoid for reduction of PVR in patients with PAH.[43] Interestingly, most patients with chronic PAH are unresponsive to an acute vasodilator challenge with short-acting agents such as epoprostenol, adenosine, or NO.[44] However, long-term administration of epoprostenol has been shown to decrease PVR in these patients despite their initial non-responsiveness.[45] Furthermore, some patients with previously severe PAH have been weaned off epoprostenol after long-term administration, with dramatically decreased PVR and improved exercise tolerance.[44] The vascular remodeling required to provide such a dramatic reduction in PVR is probably due to mechanisms besides simple local vasodilation, as predicted by Fishman in an editorial in 1998.[46] One such mechanism that appears important is the fact that long-term epoprostenol administration increases the clearance of ET-1 (a potent vasoconstrictor and mitogen). [47]
Hypoxia-induced vasoconstriction in pulmonary vessels constitutes
a fundamental difference from all other systemic blood vessels (which vasodilate
in the presence of hypoxia). Alveolar hypoxia of in vivo and in vitro whole lung,
unilateral lung, lobe, or lobule of lung all result in localized pulmonary vasoconstriction.
This phenomenon is widely referred to as "hypoxic pulmonary vasoconstriction" and
was first described nearly 60 years ago by Von Euler and Liljestrand.[48]
The HPV response is present in all mammalian species and serves as an adaptive mechanism
for diverting blood flow away from poorly ventilated to better ventilated regions
of the lung and thereby improving V̇A/![]() ratios.[49]
The HPV response is also critical for
fetal development by minimizing perfusion of the unventilated lung.
ratios.[49]
The HPV response is also critical for
fetal development by minimizing perfusion of the unventilated lung.
The HPV response occurs primarily in pulmonary arterioles of about 200-µm internal diameter in humans (60- to 700-µm internal diameter, depending on the species).[50] These vessels are advantageously situated anatomically in close relation to small bronchioles and alveoli, which permits rapid and direct detection of alveolar hypoxia. Indeed, blood may actually become oxygenated in small pulmonary arteries because of the ability of oxygen to directly diffuse across the small distance between the contiguous air spaces and vessels.[51] This direct access that gas in the airways has to small arteries makes possible a rapid and localized vascular response to changes in gas composition.
The oxygen tension at the HPV stimulus site (PsO2 ) is a function of both PAO2 and mixed venous O2 pressure (Pv̄O2 ).[52] The PsO2 -HPV response is sigmoid, with a 50% response when PAO2 , Pv̄O2 , and PsO2 are approximately 30 mm Hg. Usually, PAO2 has a much greater effect than Pv̄O2 does because O2 uptake is from the alveolar space to the blood in the small pulmonary arteries.[52]
Over the last 50 years, numerous theories were developed to explain the mechanism of HPV.[48] [53] [54] [55] Many vasoactive substances have been proposed as mediators of HPV (e.g., leukotrienes, prostaglandins, catecholamines, serotonin, histamine, angiotensin, bradykinin, and ET-1), but none has been identified as the primary mediator. In 1992, Xuan proposed that NO has a pivotal role in modulating PVR.[56] NO has involvement, but not precisely the way that Xuan first proposed. There are multiple sites of oxygen sensing with variable contributions from the NO, ET-1, and eicosanoid systems (described earlier). In vivo, HPV is currently thought to result from the synergistic action of molecules produced in both endothelial cells and smooth muscle cells.[57] However, HPV can proceed in the absence of intact endothelium, thus suggesting that the primary oxygen sensor is in the smooth muscle cell and that endothelial-derived molecules modulate only the primary HPV response.
The precise mechanism of HPV is still under investigation. However, current data support a mechanism involving the smooth muscle mitochondrial electron transport chain as the HPV sensor ( Fig. 17-9 ).[58] Additionally, reactive oxygen species (possibly H2 O2 or superoxide) are released from complex III of the electron transport chain and probably serve as second messengers to increase calcium in pulmonary artery smooth muscle cells during acute hypoxia.[59] However, alternative (less likely) mechanisms are still being investigated.[60] One alternative hypothesis suggests that smooth muscle microsomal reduced nicotinamide adenine dinucleotide phosphate (NADPH) oxido-reductase or sarcolemmal NADPH oxidase is the sensing mechanism.[60] Another, previously popular theory posited that voltage-sensitive potassium (KV ) channels were required for the HPV response. However, KV channels are no longer believed to be "obligate" but instead are thought to be attenuators because a recent study demonstrated that inhibition of KV channels failed to inhibit the HPV response.[60]
In summary, HPV is probably due to a direct action of alveolar hypoxia on pulmonary smooth muscle cells, sensed by mitochondrial ETC, with reactive oxygen species (probably H2 O2 or superoxide) serving as second messengers to increase calcium and smooth muscle vasoconstriction. The endothelial-derived products serve to both potentiate (ET-1) and attenuate (NO, PGI2 ) the HPV response. Additional mechanisms (humoral, neurogenic influences) may also modulate the baseline pulmonary vascular tone and affect the magnitude of the HPV response.
Elevated PaCO2 has a pulmonary vasoconstrictor effect. Both respiratory and metabolic acidosis will augment HPV, whereas alkalosis (both respiratory and metabolic) causes pulmonary vasodilation and serves to reduce HPV.
The clinical effects of HPV can be classified under three basic mechanisms in humans. First, life at high altitude
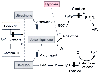 Figure 17-9
Schematic model of the mitochondrial O2
-sensing
and effector mechanism probably responsible for hypoxic pulmonary vasoconstriction
(HPV). In this model, reactive O2
species (ROS) are released from electron
transport chain complex III and act as second messengers in the hypoxia-induced calcium
(Ca2+
) increase and resultant HPV. The solid arrows
represent electron transfer steps; solid bars show
sites of electron chain inhibition. Normal mitochondrial electron transport involves
the movement of reducing equivalents generated in the Krebs cycle through complex
I or II and then through complex III (ubiquinone) and IV (cytochrome oxidase). The
Q cycle converts the dual electron transfer in complex I and II into a single electron
transfer step used in complex IV. The ubisemiquinone (a free radical) created in
this process can generate superoxide, which in the presence of superoxide dismutase
(SOD) produces H2
O2
, the probable mediator of the hypoxia-induced
increased Ca2+
and HPV. This process is amplified during hypoxia. (DPI,
diphenyleneiodonium. DPI, rotenone, and myxothiazol [not shown in figure] are inhibitors
of the proximal portion of the electron transport chain.) (From Waypa GB,
Marks JD, Mack MM, et al: Mitochondrial reactive oxygen species trigger calcium
increases during hypoxia in pulmonary artery myocytes. Circ Res 91:719–726,
2002.)
Figure 17-9
Schematic model of the mitochondrial O2
-sensing
and effector mechanism probably responsible for hypoxic pulmonary vasoconstriction
(HPV). In this model, reactive O2
species (ROS) are released from electron
transport chain complex III and act as second messengers in the hypoxia-induced calcium
(Ca2+
) increase and resultant HPV. The solid arrows
represent electron transfer steps; solid bars show
sites of electron chain inhibition. Normal mitochondrial electron transport involves
the movement of reducing equivalents generated in the Krebs cycle through complex
I or II and then through complex III (ubiquinone) and IV (cytochrome oxidase). The
Q cycle converts the dual electron transfer in complex I and II into a single electron
transfer step used in complex IV. The ubisemiquinone (a free radical) created in
this process can generate superoxide, which in the presence of superoxide dismutase
(SOD) produces H2
O2
, the probable mediator of the hypoxia-induced
increased Ca2+
and HPV. This process is amplified during hypoxia. (DPI,
diphenyleneiodonium. DPI, rotenone, and myxothiazol [not shown in figure] are inhibitors
of the proximal portion of the electron transport chain.) (From Waypa GB,
Marks JD, Mack MM, et al: Mitochondrial reactive oxygen species trigger calcium
increases during hypoxia in pulmonary artery myocytes. Circ Res 91:719–726,
2002.)
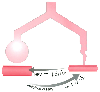 Figure 17-10
Schematic drawing of regional hypoxic pulmonary vasoconstriction
(HPV); one-lung ventilation is a common clinical example of regional HPV. HPV in
the hypoxic atelectatic lung causes redistribution of blood flow away from the hypoxic
lung to the normoxic lung, thereby diminishing the amount of shunt flow (
Figure 17-10
Schematic drawing of regional hypoxic pulmonary vasoconstriction
(HPV); one-lung ventilation is a common clinical example of regional HPV. HPV in
the hypoxic atelectatic lung causes redistribution of blood flow away from the hypoxic
lung to the normoxic lung, thereby diminishing the amount of shunt flow (![]() S/
S/![]() T)
that can occur through the hypoxic lung. Inhibition of hypoxic lung HPV causes an
increase in the amount of shunt flow through the hypoxic lung, thereby decreasing
PaO2
.
T)
that can occur through the hypoxic lung. Inhibition of hypoxic lung HPV causes an
increase in the amount of shunt flow through the hypoxic lung, thereby decreasing
PaO2
.
The three systems used to innervate the pulmonary circulation are the same ones that innervate the airways: the sympathetic, parasympathetic, and nonadrenergic noncholinergic (NANC) systems.[40]
Sympathetic (adrenergic) fibers originate from the first five thoracic nerves and enter the pulmonary vessels as branches from the cervical ganglia, as well as from a plexus of nerves arising from the trachea and main stem bronchi. These nerves act mainly on pulmonary arteries down to a diameter of 60 µm. [40] Sympathetic fibers cause pulmonary vasoconstriction through α1 -receptors. However, the pulmonary arteries also contain vasodilatory α2 -receptors and β2 -receptors. The α1 -adrenergic response predominates during sympathetic stimulation, as occurs during pain, fear, and anxiety.[40]
The parasympathetic (cholinergic) nerve fibers originate from the vagus nerve and cause pulmonary vasodilation through an NO-dependent process. [40] Binding of acetylcholine to a muscarinic (M3 ) receptor on the endothelial cell increases intracellular calcium and stimulates cNOS. [40]
NANC nerves cause pulmonary vasodilation through NO-mediated systems by using vasoactive intestinal peptide as the neurotransmitter. The functional significance of this system is still under investigation.[40]
Numerous molecules are released into the circulation that either affect pulmonary vascular tone (by binding to pulmonary endothelial receptors) or are acted on by the pulmonary endothelium and subsequently become activated or inactivated ( Table 17-2 ). The entire topic of non-respiratory function of the lung is fascinating, but beyond the scope of this chapter. Here, we will highlight the effects that circulating molecules have on pulmonary vascular tone.
Although we understand the basic effect that various circulating factors have on pulmonary vascular tone, it is unlikely that these compounds are modulators of normal pulmonary vascular tone. However, they have marked effects on pulmonary vascular tone during disease (e.g., ARDS, sepsis).
|
|
Effect of Compounds Passing through Pulmonary Circulation | ||
|---|---|---|---|
| Molecule | Activated | Unchanged | Inactivated |
| Amines |
|
Dopamine | 5-Hydroxytryptamine |
|
|
|
Epinephrine | Norepinephrine |
|
|
|
Histamine |
|
| Peptides | Angiotensin I | Angiotensin II | Bradykinin |
|
|
|
Oxytocin | Atrial natriuretic peptide |
|
|
|
Vasopressin | Endothelins |
| Eicosanoids | Arachidonic acid | PGI2 | PGD2 |
|
|
|
PGA2 | PGE1 , PGE2 |
|
|
|
|
PGF2α |
|
|
|
|
Leukotrienes |
| Purine derivatives |
|
|
Adenosine |
|
|
|
|
ATP, ADP, AMP |
| ADP, adenosine diphosphate; AMP, adenosine monophosphate; ATP, adenosine triphosphate; PG, prostaglandin. | |||
| Modified from Lumb AB: Non-respiratory functions of the lung. In Lumb AB (ed): Nunn's Applied Respiratory Physiology, 5th ed. London, Butterworths, 2000, p 309. | |||
Endogenous catecholamines (epinephrine and norepinephrine) bind to both α1 (vasoconstrictor) and β2 (vasodilator) receptors on the pulmonary endothelium but, when elaborated in high concentration, have a predominant α1 (vasoconstrictor) effect. The same is true for exogenously administered catecholamines.
Other amines (e.g., histamine, serotonin) are elaborated systemically or locally after various challenges and have variable effects on PVR. Histamine can be released from mast cells, basophils, and elsewhere. When histamine binds directly to H1 receptors on endothelium, NO-mediated vasodilation occurs (as seen after epinephrine-induced pulmonary vasoconstriction). Direct stimulation of H2 receptors on smooth muscle cell membranes also causes vasodilation. In contrast, stimulation of H1 receptors on the smooth muscle membrane results in vasoconstriction. Serotonin (5-hydroxytryptamine [5-HT]) is a potent vasoconstrictor that can be elaborated from activated platelets (e.g., after pulmonary embolism) and lead to acute severe pulmonary hypertension.[67]
Numerous peptides circulate and cause either pulmonary vasodilation (e.g., substance P, bradykinin, and vasopressin [a systemic vasoconstrictor]) or vasoconstriction (e.g., neurokinin A and angiotensin). These peptides produce clinically detectable effects on PVR only when administered in high concentration (e.g., exogenous administration or in disease).
Two other classes of molecules must be mentioned for completeness, eicosanoids (vasoactive effects discussed earlier) and purine nucleosides (which are similarly highly vasoactive).[40] Adenosine is a pulmonary vasodilator in normal subjects, whereas adenosine triphosphate (ATP) has a variable "normalizing effect," depending on baseline pulmonary vascular tone. [68]
Blood can use several possible pathways to travel from the right side of the heart to the left without being fully
Several right-to-left blood flow pathways through the lungs and heart do not pass by or involve the alveoli at all. The bronchial and pleural circulations originate from systemic arteries and empty into the left side of the heart without being oxygenated; these circulations constitute the 1% to 3% true right-to-left shunt normally present. With chronic bronchitis, the bronchial circulation may carry 10% of the cardiac output, and with pleuritis, the pleural circulation may carry 5% of the cardiac output. Consequently, as much as a 10% and a 5% obligatory right-to-left shunt may be present, respectively, under these conditions. Intrapulmonary arteriovenous anastomoses are normally closed, but in the face of acute pulmonary hypertension, such as may be caused by a pulmonary embolus, they may open and result in a direct increase in right-to-left shunting. The foramen ovale is patent in 20% to 30% of individuals, but it normally remains functionally closed because left atrial pressure exceeds right atrial pressure. However, any condition that causes right atrial pressure to be greater than left atrial pressure may produce a right-to-left shunt, with resultant hypoxemia and possible paradoxical embolization. Such conditions include the use of high levels of PEEP, pulmonary embolization, pulmonary hypertension, chronic obstructive pulmonary disease, pulmonary valvular stenosis, congestive heart failure, and postpneumonectomy states.[69] Indeed, even such common events as mechanical ventilation[70] and reaction to the presence of an endotracheal tube during the excitement phase of emergence from anesthesia [71] have caused right-to-left shunting across a patent foramen ovale and severe arterial desaturation (with the potential for paradoxical embolization).[71]
Transesophageal echocardiography (TEE) has been demonstrated to be the most sensitive modality for diagnosing a patent foramen ovale in anesthetized patients with elevated right atrial pressure.[72] Esophageal to mediastinal to bronchial to pulmonary vein pathways have been described and may explain in part the hypoxemia associated with portal hypertension and cirrhosis. There are no known conditions that selectively increase thebesian channel blood flow (thebesian vessels nourish the left ventricular myocardium and originate and empty into the left side of the heart).
|
|
|
|
|
|
|
|
|
|
|
|
|